
Bio-degradable cards
Every card imaginable!
|
Bio-degradable cards Every card imaginable! Cystic Fibrosis Symptoms Causes Diagnosis Treatment
DisclaimerThis guide is for informational purposes only and is not a substitute for professional medical advice, diagnosis, or treatment. Always consult a qualified healthcare provider for any personal health concerns or questions about cystic fibrosis. Dated: December 30, 2025. Table of ContentsUnderstanding Cystic Fibrosis: What It Is and How It Affects the BodyCystic fibrosis is a lifelong genetic condition that primarily affects the lungs and digestive system. It happens when a faulty gene causes mucus in the body to become unusually thick and sticky. This buildup can clog airways, trap bacteria leading to infections, and block important digestive enzymes from doing their job. In the UK, more than 11,300 people live with cystic fibrosis, and thanks to newborn screening introduced across the country in 2007, most cases are spotted early. It's completely understandable if a new diagnosis feels overwhelming—many parents and individuals go through a mix of emotions at first. The good news is that treatments have advanced hugely, and people with cystic fibrosis are living longer, fuller lives than ever before, with recent data showing that half of those born today are predicted to live to at least 66 years. What Causes Cystic Fibrosis?At its core, cystic fibrosis is caused by mutations in a gene called the CFTR gene. This gene provides instructions for making a protein called the cystic fibrosis transmembrane conductance regulator (CFTR).
This thick mucus disrupts normal function in organs lined with epithelial cells, leading to the main challenges of cystic fibrosis. There are over 2,000 known mutations, but the most common in the UK is F508del (also written as ΔF508), carried by around 90% of people with cystic fibrosis on at least one copy of their genes. Experiences can vary depending on the specific mutations involved. How Cystic Fibrosis Affects the LungsThe lungs are often the most noticeably affected part of the body in cystic fibrosis, as the thick mucus makes everyday breathing more challenging.
How Cystic Fibrosis Affects the Digestive SystemMany people with cystic fibrosis experience digestive issues because the same thick mucus affects the pancreas and intestines.
Effects on Other Parts of the BodyCystic fibrosis doesn't stop at the lungs and gut—it can influence several other systems, though not everyone experiences these to the same degree.
Everyone's experience with cystic fibrosis is unique, influenced by their gene mutations, early care, and access to treatments. While it's a serious condition, proactive management and recent breakthroughs mean many people with cystic fibrosis work, study, travel, and build families. If you're supporting someone with cystic fibrosis or living with it yourself, reaching out to specialist teams early makes a real difference. The outlook continues to improve, offering genuine hope for the future. 
Diagnosis and Early Signs: Spotting the ConditionIn the UK, most babies with cystic fibrosis are identified very early through routine newborn screening, which has been offered to all newborns since 2007. This simple heel-prick blood test, done around day five of life, checks for several conditions including cystic fibrosis by measuring levels of a substance called immunoreactive trypsinogen (IRT). If levels are raised, further genetic testing on the same blood sample looks for common mutations. It's completely understandable to feel anxious if you're contacted about an abnormal result—many parents do—but remember that not all positive screens lead to a diagnosis, and early detection allows treatment to start straight away, which makes a huge difference to health outcomes. Thanks to this national programme, the vast majority of people with cystic fibrosis are diagnosed in the first few weeks of life. Recent UK Cystic Fibrosis Registry data shows that the median age at diagnosis for those under 16 is just 21 days. Starting care early helps prevent complications and supports better growth and lung health from the outset. The Newborn Screening ProcessThe heel-prick test is quick and part of standard care for every baby in the UK.
Confirming the Diagnosis: The Sweat TestThe sweat test remains the gold standard for diagnosing cystic fibrosis and is painless and reliable.
Early Signs in Babies and Young ChildrenEven with screening, some parents notice clues before or alongside the process, though symptoms vary widely.
If these signs ring true, speak to your GP or health visitor promptly—they can arrange referrals to rule out or confirm cystic fibrosis. Signs in Older Children, Teenagers, and AdultsWhile rare now due to screening, some people with milder forms of cystic fibrosis are diagnosed later in life.
No matter the age, if symptoms suggest cystic fibrosis, your GP can refer you to a specialist centre for sweat testing and genetic analysis. The Emotional Impact of DiagnosisReceiving a diagnosis of cystic fibrosis can bring a whirlwind of emotions—shock, worry, grief, or even relief at finally having answers. It's normal for parents to feel overwhelmed thinking about the future, or for adults to grapple with adapting established lives to new treatments. Many families describe an initial sense of isolation, but connecting with specialist teams early provides practical and emotional support. Support groups and charities like the Cystic Fibrosis Trust offer spaces to talk with others who've been through similar experiences, which can ease that burden considerably. The key reassurance is that early diagnosis and modern care transform prospects. Babies identified through screening often start treatments before symptoms become severe, leading to healthier childhoods. For those diagnosed later, catching it sooner rather than later still opens doors to effective management. Specialist cystic fibrosis teams across the UK are there to guide you every step of the way, tailoring care to individual needs and offering hope grounded in real progress. 
Daily Management and Treatment OptionsManaging cystic fibrosis day to day revolves around a personalised routine that helps keep lungs clear, prevents infections, supports digestion, and maintains overall health. This routine is developed with input from a specialist multidisciplinary team at a cystic fibrosis centre, and it often evolves over time as needs change. Many people find that what feels overwhelming at first gradually becomes a natural part of life, allowing them to focus on work, family, and hobbies. It's completely understandable if the daily commitments seem daunting initially—most families and individuals feel that way—but with support from the team, it becomes more manageable and empowering. In the UK, care follows NICE guidelines and is delivered through NHS specialist centres, where teams review progress regularly and adjust plans based on annual assessments and any changes in health. Airway Clearance TechniquesClearing thick mucus from the lungs is a cornerstone of daily management for almost everyone with cystic fibrosis, usually done at least once or twice a day, often more during infections.
Physical exercise also acts as a natural airway clearance method, improving lung function and overall fitness—many incorporate activities like walking, swimming, or cycling into their day. Medications for Lungs and AirwaysA range of inhaled and oral medicines help thin mucus, open airways, and fight or prevent infections.
CFTR Modulator TherapiesThese groundbreaking medicines target the underlying faulty CFTR protein, representing the biggest advance in cystic fibrosis care in recent decades. Availability on the NHS has expanded significantly, with around 95% of people in England now eligible.
Eligibility is determined by genetic testing, and modulators are often combined with existing treatments—many notice fewer exacerbations and more energy. Nutrition and Digestive SupportGood nutrition is vital to support growth, immunity, and energy levels, with most needing a high-calorie diet.
Monitoring and Preventing ComplicationsRegular clinic visits, home monitoring (like spirometry), and annual reviews catch issues early.
While the routine requires commitment, it helps maintain stability and take advantage of improving outcomes—recent registry data shows many living well into adulthood and beyond. Research continues to bring new options, providing real grounds for optimism. 
Living Well: Nutrition, Exercise, and Emotional SupportLiving well with cystic fibrosis means finding practical ways to support your body and mind through good nutrition, regular movement, and emotional care. These elements work together to help maintain energy levels, strengthen lungs, support growth, and build resilience against the daily challenges of the condition. Many people with cystic fibrosis lead active, fulfilling lives—pursuing education, careers, relationships, and travel—thanks to personalised strategies developed with their specialist team. It's completely understandable if balancing these feels tricky at times, especially with treatments on top, but small, consistent steps often make a big difference, and your cystic fibrosis centre is there to guide you every step of the way. In the UK, specialist dietitians, physiotherapists, and psychologists are integral parts of NHS cystic fibrosis teams, offering tailored advice based on guidelines from the Cystic Fibrosis Trust and NICE. Nutrition: Fuelling Your Body EffectivelyGood nutrition is essential for everyone with cystic fibrosis because malabsorption can make it harder to get enough energy and nutrients from food, even when eating well. Most people need a higher-calorie intake—often 20-50% more than someone without the condition—to support lung health, immunity, and daily activities.
If weight gain is a challenge, options like overnight tube feeds can provide gentle extra support. Working closely with your dietitian helps create enjoyable meal plans that fit your tastes and lifestyle, turning nutrition into a positive part of daily life. Exercise: Building Strength and Clearing AirwaysRegular physical activity is one of the best ways to support lung function, clear mucus naturally, improve fitness, and enhance overall wellbeing in cystic fibrosis. It complements airway clearance techniques and can reduce the risk of infections while boosting mood and energy.
Evidence shows that staying active correlates with better lung function and fewer hospital stays. Many people with cystic fibrosis compete in sports, run marathons, or simply enjoy family walks, proving it can fit seamlessly into life. Emotional and Psychological SupportThe demands of cystic fibrosis—treatments, appointments, and uncertainties—can sometimes affect mood, relationships, or self-esteem, and it's normal to experience anxiety, low moments, or frustration along the way. Prioritising mental health is just as important as physical care, and seeking help early is a sign of strength.
With advances in care, including modulators, the outlook is brighter than ever—recent UK Registry data predicts that half of babies born today will live to at least 66 years, and many adults thrive well beyond previous expectations. By nurturing nutrition, embracing movement, and looking after your emotional health, you're investing in a fuller, more vibrant life. Your multidisciplinary team is equipped to help tailor these approaches, offering ongoing encouragement and adjustments to keep things positive and achievable. 
Potential Complications and Long-Term OutlookEven with excellent daily management, cystic fibrosis can sometimes lead to complications that require close monitoring and additional care. These issues often develop gradually, and regular check-ups with your specialist team help spot them early, allowing for timely adjustments to keep things stable. It's natural to feel some uncertainty when thinking about potential longer-term challenges—many parents and individuals do—but the focus on proactive care means most people experience fewer severe problems than in previous generations, thanks to advances in treatments like modulators. In the UK, NHS cystic fibrosis centres follow national guidelines to screen for and manage these complications, often catching them before they significantly impact daily life. Respiratory ComplicationsThe lungs remain the area most affected by cystic fibrosis over time, as repeated infections and inflammation can cause gradual changes.
Cystic Fibrosis-Related DiabetesDamage to the pancreas from thick secretions can affect insulin production, leading to cystic fibrosis-related diabetes (CFRD), a unique form distinct from type 1 or type 2 diabetes.
Liver and Digestive ComplicationsThick bile can block ducts in the liver and gallbladder, while intestinal issues arise from sticky mucus.
Bone and Joint HealthMalabsorption of vitamins and minerals, along with inflammation, steroids, and reduced activity, can impact bones.
Other Considerations
The Long-Term OutlookThe future for people with cystic fibrosis is brighter than ever, with ongoing research and treatments transforming prospects. Recent data from the UK Cystic Fibrosis Registry shows that half of babies born today are predicted to live to at least 66 years, a remarkable shift driven by newborn screening, specialist care, and widespread access to CFTR modulators. Early diagnosis, personalised management, and breakthroughs mean many adults now work full-time, travel, and build families well into their 40s, 50s, and beyond. While cystic fibrosis remains a serious condition requiring lifelong commitment, the emphasis on prevention and innovation offers genuine hope. Your specialist team will discuss your individual outlook based on regular assessments, helping you plan with confidence and focus on living fully today. 
Help and Further ResourcesFinding the right support can make a big difference when living with or caring for someone with cystic fibrosis. There are several trusted organisations and services across the UK ready to offer practical advice, emotional support, and reliable information. Connecting with them often helps reduce feelings of isolation and provides reassurance during challenging times. National Support: Cystic Fibrosis TrustThe Cystic Fibrosis Trust is the main UK-wide charity dedicated to cystic fibrosis, supporting everyone affected by the condition through high-quality resources and services.
NHS ResourcesThe NHS provides clear, official information for people in the UK. Visit www.nhs.uk/conditions/cystic-fibrosis for straightforward overviews of symptoms, treatments, and living with the condition, plus links to finding your local specialist cystic fibrosis centre. Your GP or specialist team can refer you directly to multidisciplinary NHS cystic fibrosis services, which include ongoing monitoring and coordinated care. Medical Identification CardsMany people with cystic fibrosis find it helpful to carry a medical identification card, which can quickly alert others to the condition in emergencies or everyday situations where extra understanding is needed. Our medical cards provide key details about cystic fibrosis. They’re discreet, durable, and designed specifically with the needs of people with cystic fibrosis in mind. You can find out more on our website. Other Ways to ConnectOnline communities, local support groups through your cystic fibrosis centre, or parent networks can also be lifelines. Your specialist team can often point you towards relevant options in your area. Reaching out for support is a positive step, and many families and individuals find that these resources help them feel more confident and connected. Whether it's a quick question to a helpline or ongoing peer support, there's help available whenever you need it. 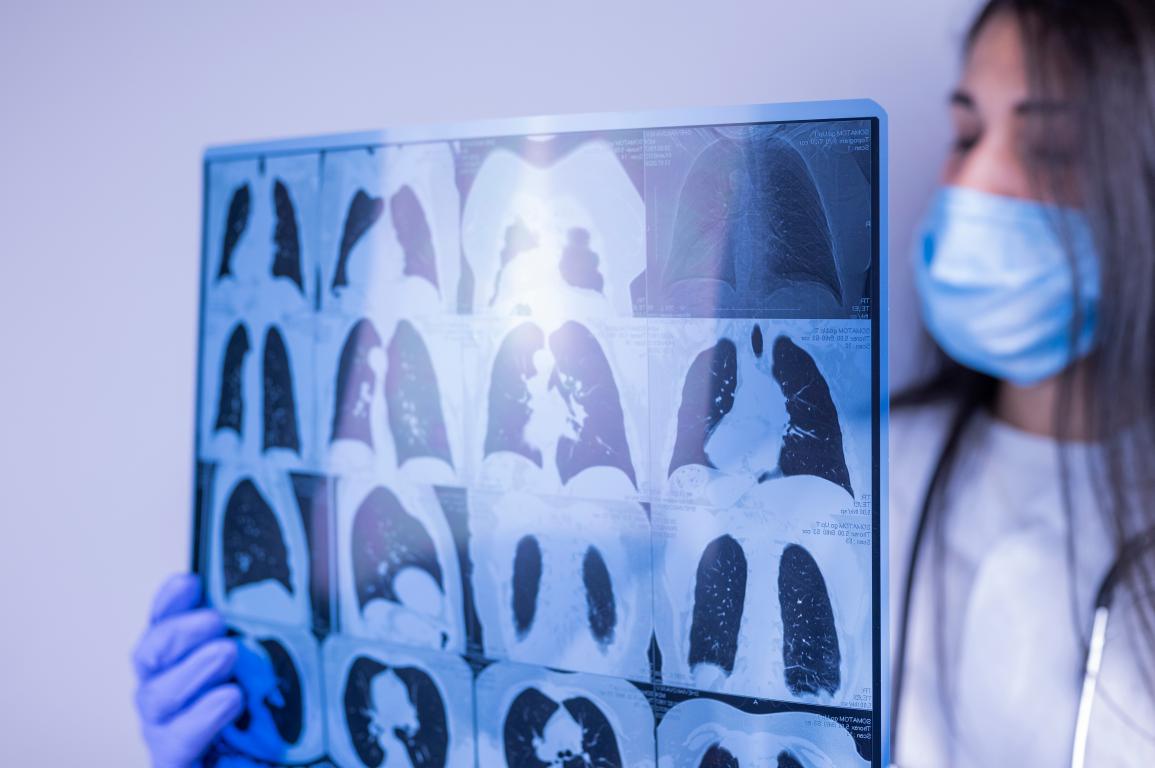
FAQHere are answers to some of the most common questions about cystic fibrosis. This section provides clear, straightforward information based on current understanding and UK care practices. For personalised guidance, your specialist team is the best source. What causes cystic fibrosis?Cystic fibrosis is a genetic condition passed down when both parents carry a faulty CFTR gene. Each pregnancy has:
It's not contagious. Carrier testing via blood or saliva can help with family planning, including options like IVF with pre-implantation genetic diagnosis. Over 2,000 mutations exist, with F508del being the most common in the UK. How is cystic fibrosis diagnosed in newborns?Newborns in the UK are screened via the heel-prick test around day five. This checks immunoreactive trypsinogen (IRT) levels, and if raised, genetic analysis is done. A positive screen is confirmed with a sweat test, which measures salt levels. Early diagnosis allows treatment to start immediately, improving growth, lung health, and long-term outcomes. Can people with cystic fibrosis have children?Yes, though fertility issues are common. Most men have congenital absence of the vas deferens, but sperm retrieval and ICSI often succeed. Women usually have normal fertility, though thicker cervical mucus can make conception harder, and pregnancy requires closer monitoring. With specialist support, many people have healthy pregnancies and children. Is exercise safe for someone with cystic fibrosis?Exercise is strongly recommended. It helps clear mucus, strengthens lungs and muscles, boosts fitness, and supports mental health. Activities can be adapted to all levels, with physiotherapist guidance on suitable options, hydration, and salt intake. What about school or work with cystic fibrosis?Children and adults usually attend school or work full-time. Accommodations may include:
Open communication with teachers or employers ensures routines run smoothly. Are there new treatments on the horizon?Yes. CFTR modulators have transformed care, and ongoing research is exploring gene therapies, advanced correctors, and other innovations. The Cystic Fibrosis Trust provides trial updates and participation opportunities. How does cystic fibrosis affect mental health?Cystic fibrosis can cause anxiety, stress, or feelings of difference. Talking therapies, peer support, and counselling at cystic fibrosis centres help build coping strategies. Early attention prevents problems from escalating. Can cystic fibrosis be cured?There is currently no cure, but treatments manage symptoms effectively. Life expectancy and quality of life have improved dramatically thanks to early diagnosis, better care, and new therapies. Research continues into potential cures, including gene editing. What should I eat with cystic fibrosis?A high-calorie, high-fat diet is usually needed, alongside pancreatic enzymes. Include foods like full-fat dairy, nuts, avocados, and added oils or butter, with prescribed vitamins. Dietitians personalise plans for tastes and lifestyle. Hydration and extra salt support overall health. How often are check-ups needed?Specialist centre visits usually occur every 3 months, covering lung function, sputum and blood tests, and nutrition and wellbeing reviews. Frequency may increase during illness. Annual assessments provide comprehensive overviews. Does cystic fibrosis affect life expectancy?Life expectancy has improved greatly. Recent UK data predicts half of babies born today will live to at least 66 years, with many adults thriving into their 50s and beyond. Early care and treatment advances continue to improve lifespan and quality of life. 
ConclusionThis guide provides practical information for living with cystic fibrosis in the UK. It covers understanding the condition, early diagnosis via newborn screening, daily management, nutrition, exercise, and potential complications. Consistent care and engagement with your specialist team make a real difference. Modern treatments, particularly CFTR modulators available on the NHS, have significantly improved outcomes, helping people stay healthier for longer and enjoy active lives. While cystic fibrosis is lifelong, early intervention, personalised support from specialist centres, and access to resources like the Cystic Fibrosis Trust improve the outlook. You are not alone—NHS care and support organisations are available every step of the way. This guide is for general information and does not replace professional medical advice. Always discuss your individual situation with your healthcare team for recommendations tailored to your needs. Staying connected with your specialist team and trusted support networks helps you manage cystic fibrosis confidently and live well. This information is accurate as of December 2025. Medical knowledge and treatments evolve, so consult your healthcare professional for the latest advice.
© 2024 The Card Project Uk Ltd
VAT: 453 2087 06
|